
尤其对于肾脏病患者,增龄与肾脏病本身互相影响,更容易加重肾脏病的进展。如何优雅面对增龄做到健康老龄化,去乙酰化酶,也称为长寿蛋白的Sirtuins一直是人们的期望。随着研究不断深入,Sirtuins在肾脏健康和疾病中的作用也逐渐揭开神秘的面纱。相信刚刚发表在肾脏病经典综述杂志《NatureReviewsNephrology》上的这篇“肾脏健康与疾病中的去乙酰化酶”能给您的龙年新年带来不一样的认知礼物。借此佳节之际,小编也祝您和家人龙年新年快乐,身体健康,平安喜乐,阖家幸福!
:10.1038/s46-4.
去乙酰化酶(Sirtuins,SIRTs)在模式生物中是公认的长寿调节因子。自从最初发现SIRTs可以在线虫和苍蝇中延长寿命,对这些蛋白质特性的鉴定,使人们了解了它们作为精细传感器将代谢活性与氧化状态联系起来的作用。SIRTs在肾脏发育和生理功能的重要的生物过程扮演主要角色,包括线粒体代谢、氧化应激、自噬、DNA修复和炎症。此外,SIRT活性的改变与急慢性肾脏疾病的病理生理学和进展有关,包括急性肾损伤、糖尿病肾病、慢性肾脏疾病、多囊肾病、自身免疫性疾病和肾脏衰老。
SIRTs在这些疾病中的肾脏保护作用让它们成为了更有吸引力治疗靶点。大量SIRT激活化合物在肾脏疾病模型中显示出有益作用;但是,尚需进一步研究治疗和/或预防肾脏疾病和增加人类平均寿命的新型SIRT靶向蛋白策略。
要点
Sirtuins(SIRTs)最初由于其在实验模型中调节寿命的假定作用而引起了极大的科学兴趣;然而,现在人们普遍认为它们是健康老龄化的驱动因素。
SIRTs调节细胞内稳态的关键过程,包括代谢、线粒体功能、氧化应激、DNA修复、凋亡、衰老和炎症。
SIRTs在肾脏发育和生理过程中发挥重要作用,因为其对高度特化和分化的肾细胞类型(包括足细胞和肾小管细胞)具有广泛的细胞保护作用。
SIRT改变已被确定为人类和实验模型中各种肾脏疾病的驱动因素,包括急性肾损伤、慢性肾脏疾病和糖尿病肾脏疾病。
sirtuin活性化合物的鉴定是该领域的主要目标;在实验模型中一些sirtuin激活化合物已被证明增强SIRT活性并给予肾脏保护,但很少有人进入临床试验。
为了认识到这些干预措施在治疗和预防肾脏疾病以及增加人类健康寿命方面的潜力,需要具有改进效力和生物利用度的新型SIRT靶向化合物。
引言
Sirtuins(SIRTs)是一类进化上保守的组蛋白去乙酰化酶(histonedeacetylases,HDACs),其活性需要烟酰胺腺嘌呤二核苷酸(nicotinamideadeninedinucleotide,NAD+)。自30年前首次发现以来,这一家族的蛋白质一直是广泛研究的焦点,主要是因为它们在模型生物中的寿命调节中起着假定的作用,包括酿酒酵母、秀丽隐杆线虫和黑腹果蝇。此外,至少有一个SIRT,SIRT6,与雄性小鼠的寿命有关。然而,随着后续研究对SIRTs影响线虫和昆虫寿命的概念提出挑战,最初对SIRTs作为长寿蛋白的热情减弱。
随着关于SIRTs在长寿中作用的争论展开,大量研究表明,SIRTs是平衡包括肾脏在内的代谢活跃器官中过多应激源的关键介质。这些发现导致了SIRTs是健康老龄化的重要驱动因素,而不是长寿调节因子的概念。例如,脑特异性Sirt1在小鼠中的过度表达可显著延缓衰老。相反,Sirt3或Sirt6的缺失会导致小鼠的早衰相关器官损害。
本文就SIRTs在调节肾脏发育及细胞能量稳态、抗氧化活性和DNA修复等生理过程中的作用作一综述。我们还讨论了SIRTs在急慢性肾脏疾病发病机制中重要作用的分子机制,重点讨论了最常研究的SIRTs,SIRT1,SIRT3和SIRT6。最后,我们强调了SIRTs作为拮抗人类肾脏疾病和肾脏衰老的治疗靶点的潜力,以及确定刺激这些蛋白质的新策略的必要性。
哺乳动物sirtuins
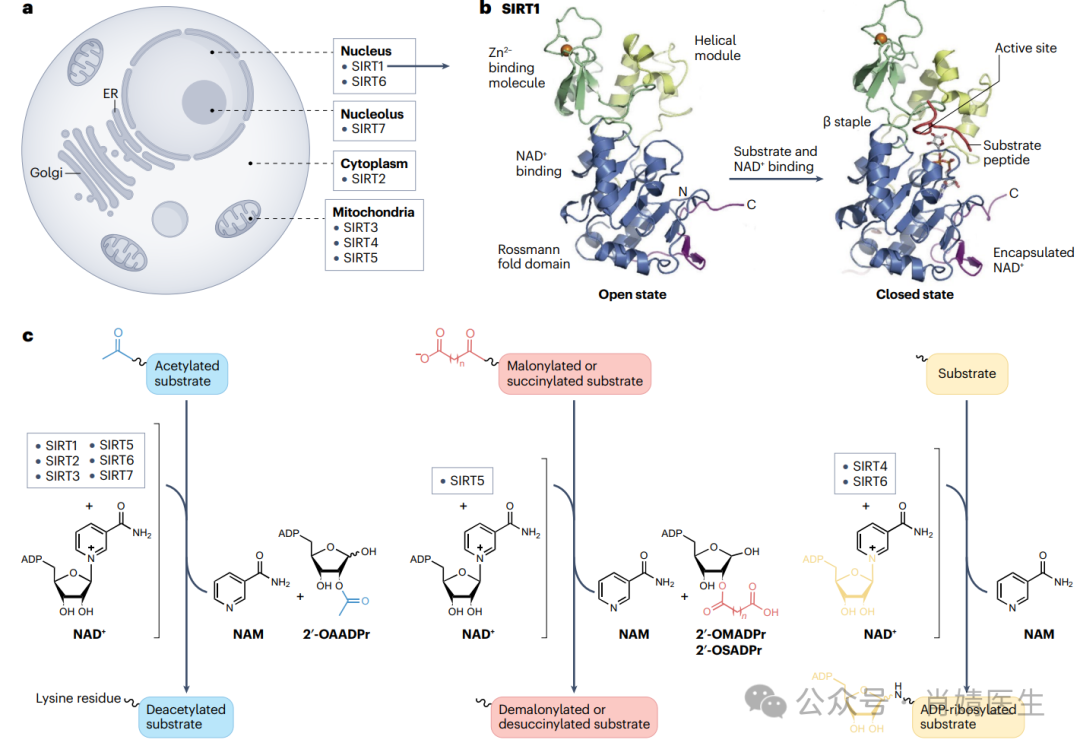
所有SIRT均包含约270个残基催化核心以及长度和序列不同的N端和C端片段。哺乳动物SIRT作用于不同的细胞区,其中它们对广泛的酰基蛋白底物表现出特异性(图1)。
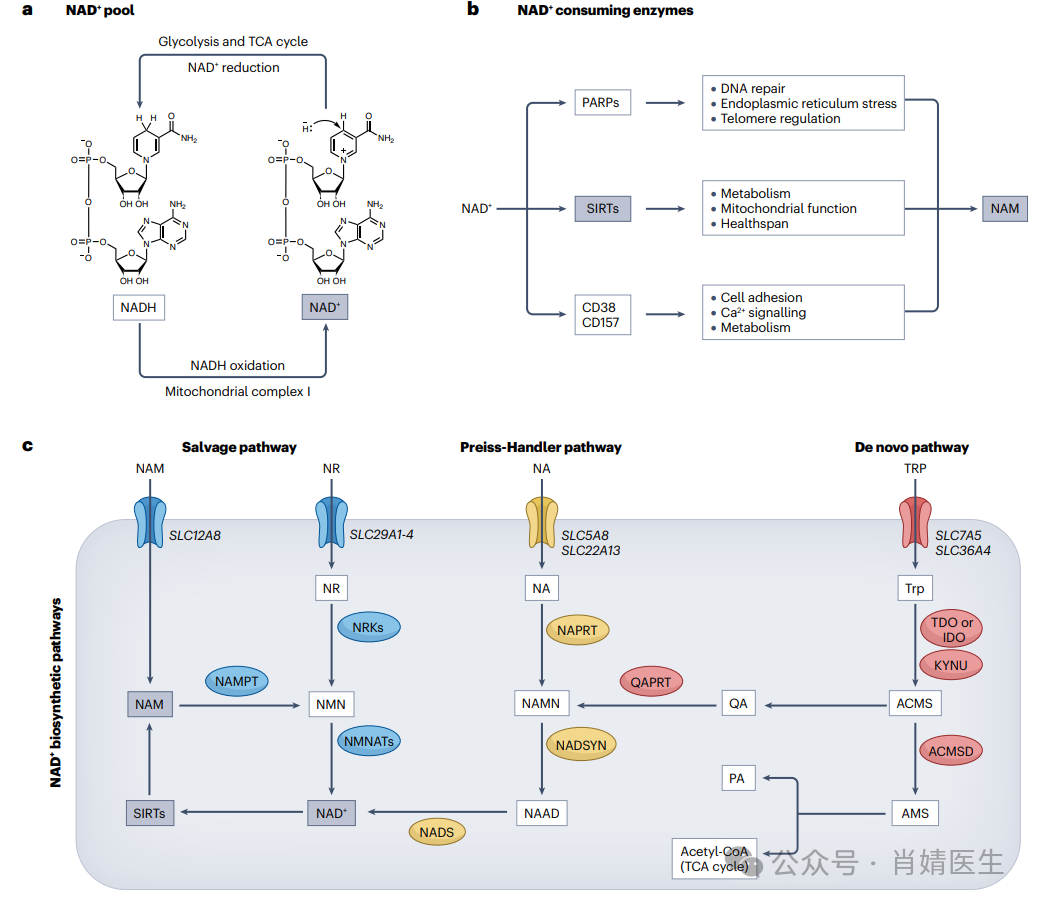
图1|SIRT定位、结构和酶活性。a、在真核细胞中,SIRT1和SIRT6主要定位于细胞核,而SIRT7仅限于核仁区。SIRT2位于细胞质中,SIRT3、SIRT4和SIRT5定位于线粒体基质。b,所有sirtuins都有一个保守的催化核心,由烟酰胺腺嘌呤核苷酸(NAD+)结合的罗斯曼折叠、Zn2+结合模块和包含NAD+结合环的螺旋模块组成。这里以SIRT1的分子结构为例。目标蛋白的酰基赖氨酸残基和NAD+从对侧插入裂隙内的疏水通道。插入后,分子从开放形式转变为封闭形式,使赖氨酸的乙酰化ε-氨基与NAD+的核糖部分相邻。酰基肽的主链与SIRT活性位点的三链反平行β-短纤维形成氢键,使其脱酰基而无需序列保守性。c、SIRT1、SIRT2、SIRT3、SIRT5、SIRT6和SIRT7具有赖氨酸脱乙酰化活性。辅酶NAD+作为目标蛋白赖氨酸残基乙酰基的受体,形成2′-O-乙酰基-ADP-核糖(2′-OAADPr)和烟酰胺(NAM)。SIRT5还从靶蛋白的赖氨酸中去除丙二酰或琥珀酰基团,将其转移到NAD+,分别生成2′-O-丙二酰-ADP-核糖(2′-OMADP)和2′-O-琥珀酰-ADP-核糖(2′-OSADP)以及NAM。SIRT4和SIRT6具有ADP-核糖转移酶活性,以NAD+为ADP-核糖基团的供体,产生NAM。b部分经参考文献Elsevier许可改编。
除SIRT4外,所有SIRT均表现出通过两个连续步骤进行的脱乙酰酶活性;NAD+最初被切割成烟酰胺(nicotinamide,NAM)和ADP核糖,随后作为目标蛋白赖氨酸乙酰基的受体,生成O-乙酰-ADP核糖(2′-O-malonyl-ADP-ribose,2′-OAADPr)。SIRT5具有额外的酰化酶活性,并将目标蛋白的赖氨酸中的丙二酰或琥珀酰基团转移到ADP核糖,分别生成2′-O-丙二酰-ADP核糖(2′-O-malonyl-ADP-ribose,2′-OMADP)和2′-O-琥珀酰-ADP核糖(2′-O-succinyl-ADP-ribose,2′-OSADP)。SIRT4具有ADP-核糖转移酶活性,促进ADP-核糖从NAD+转移到靶蛋白的赖氨酸,产生NAM。细胞的代谢状态决定NAD+水平和SIRT酶活性(图2)。
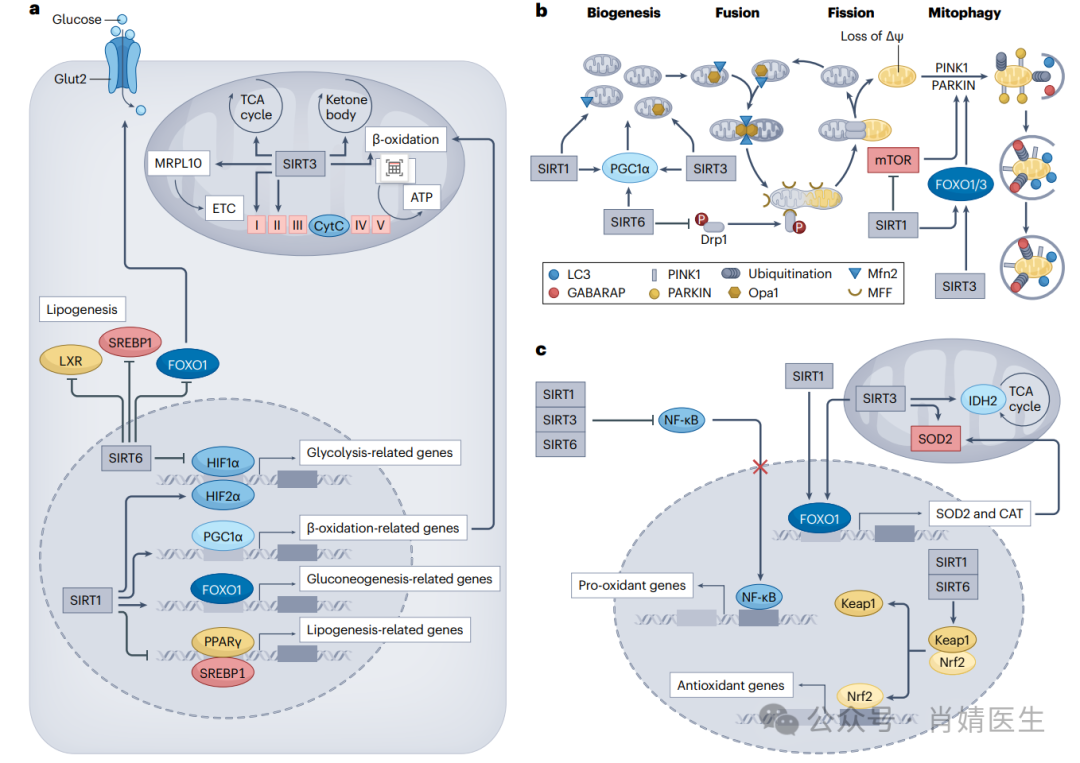
图2|NAD+功能和生物合成途径。a、在哺乳动物细胞中,NAD+在氧化还原对NAD+和NADH之间稳定循环。NAD+通过糖酵解途径和三羧酸(TCA)循环中的脱氢酶还原为NADH,而NADH氧化为NAD+发生在线粒体氧化磷酸化的第一步,由复合物介导,包括SIRTs(SIRTs)、poly(ADP-核糖)聚合酶(PARPs)和cADP-核糖合成酶(如CD38和CD157),在酶促反应过程中积极消耗NAD+。这些NAD+消耗酶产生副产物烟酰胺(NAM)。c、NAM也可以通过特定的转运体被细胞吸收,如小肠中的SLC12A8。为了补充NAD+水平,补救途径通过两种酶反应将细胞内NAM转化为NAD+。烟酰胺磷酸核糖转移酶(NAMPT)将NAM转化为烟酰胺单核苷酸(NMN),随后通过烟酰胺单核苷酸腺苷酸转移酶1–3(NMNATs)转化为NAD+。在这一途径中,NMN也可由烟酰胺核糖苷激酶1和2(NRK)产生,后者通过特异性转运体(SLC29A1-4)转化导入细胞的烟酰胺核糖苷(NR)。NAD+也可以通过Preiss–Handler和从头途径生成。在Preiss–Handler途径中,膳食烟酸(NA)通过SLC5A8或SLC22A13转运蛋白进入细胞,并通过烟酸磷酸核糖转移酶(NAPRT)转化为烟酸单核苷酸(NAMN)。NAD合成酶(NADSYN)将NAMN转化为烟酸腺嘌呤二核苷酸(NAAD),NAAD通过NAD+合成酶(NADS)转化为NAD+。在从头途径中,膳食色氨酸(Trp)通过SLC7A5和SLC36A4转运蛋白进入细胞。色氨酸2,3-双加氧酶(TDO)、吲哚胺2,3-双加氧酶(IDO)和犬尿氨酸酶(KYNU)等多种酶通过多种反应将色氨酸转化为α-氨基-β-羧基黏液酸-ε-半醛(ACMS)。ACMS发生自发环化,生成喹啉酸(QA),通过喹啉酸磷酸核糖转移酶(QPRT)将其转化为NAMN。ACMS还可以通过ACMS脱羧酶(ACMSD)转化为α-氨基-β-黏蛋白-ε-半醛(AMS)。AMS用于TCA循环或非酶转化为吡啶甲酸(PA)。
核sirtuins
三个SIRT1、SIRT6和SIRT7定位于细胞核。最常研究的核SIRT是SIRT1,它使H4赖氨酸16(H4lysine16,H4K16)和H3赖氨酸9(H3lysine9,H3K9)处的组蛋白去乙酰化,从而调控基因转录。还报道了SIRT1的胞质定位。细胞质SIRT1与非组蛋白的去乙酰化相关,包括DNA结合转录因子及其协同调节分子,进一步扩大了该蛋白的生物学作用。
SIRT6直接去乙酰化组蛋白H3K9和H3K56,并在维持基因组稳定性和端粒功能方面发挥重要作用。SIRT7的目标大部分未被探测。SIRT7主要定位于核仁,与RNA聚合酶I机制相关,是核糖体DNA转录所必需。一个可能在复制性衰老中起作用的SIRT7细胞质池也已被鉴定。
胞质sirtuins
胞质SIRT2与微管相互作用,使α微管的赖氨酸40脱乙酰化。在大多数体细胞中,乙酰化微管蛋白是有丝分裂所必需的,并定位于参与细胞周期各阶段的细胞器。SIRT2限制微管蛋白乙酰化的能力在有丝分裂的调节中发挥作用。特别是,SIRT2在G2/M期上调,在M期抑制,以促进细胞周期中的有丝分裂退出。SIRT2抑制的失败延长了M期。
线粒体sirtuins
SIRT3已被描述为线粒体整体蛋白脱乙酰化的主要调节器,根据最初的研究发现,SIRT3缺陷小鼠的线粒体蛋白乙酰化水平较高,而Sirt4和Sirt5缺陷小鼠的线粒体蛋白乙酰化水平不高。SIRT3也可在细胞核中发现,其靶向组蛋白H3和H4。在应激反应中,核SIRT3可以转移到线粒体,这表明它可能是防御细胞损伤机制的关键调节因子。
在线粒体内,SIRT4主要是一种ADP核糖基酶,与其他SIRTs不同,它在热量限制条件下失活。SIRT5最初被鉴定为线粒体脱乙酰酶,但精氨酸(Arg105)和酪氨酸(Tyr102)残基在其活性部位的存在赋予额外的去克隆酶和去琥珀酸酶活性。
哺乳动物生理中的Sirtuins
SIRTs的初步研究主要集中在确定其酶活性的特定底物,目的是揭示其对各种细胞过程的独特贡献。随着对SIRTs的理解不断发展,新出现的范例表明,SIRTs调控靶蛋白的功能簇,协调精细调节的生理反应,跨越多种细胞过程,包括代谢调节、氧化应激、细胞存活和保持基因组稳定性。它们参与多方面的生物途径,建立了SIRTs作为细胞内环境稳定的关键调节因子。
代谢调节
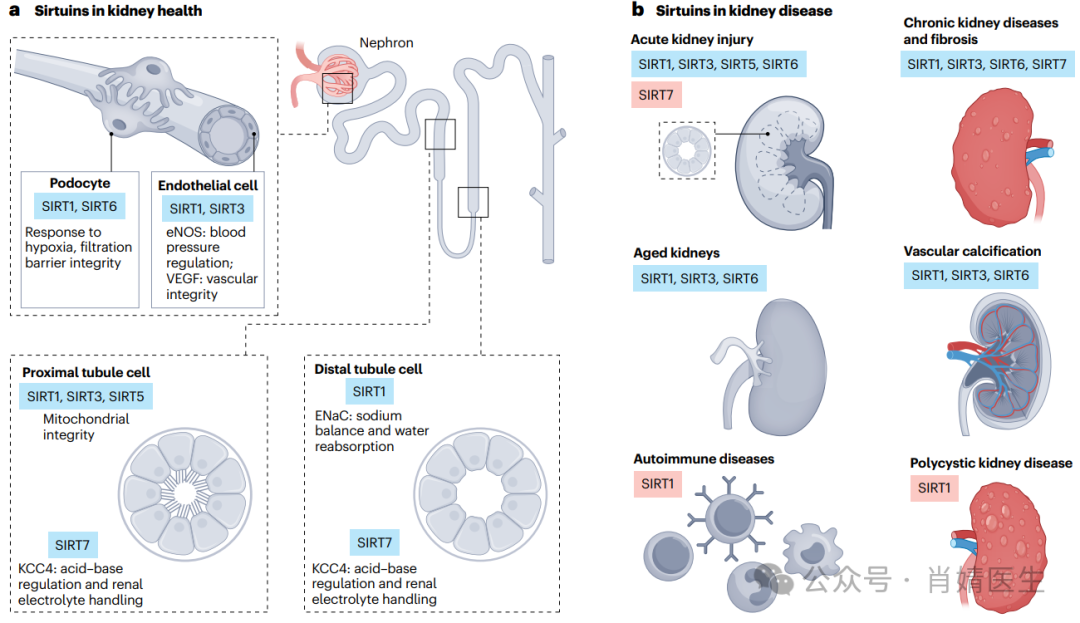
图3|sirtuins调控的主要生物过程概述。a、不同的sirtuins(SIRTs)在不同程度上调节细胞代谢。在细胞核中,SIRT1激活缺氧诱导因子2(HIF2α)、过氧化物酶体增殖物激活受体γ辅激活因子1-α(PGC1α)和叉头盒O型转录因子1(FOXO1),进而分别激活糖酵解、β-氧化和糖异生相关基因的转录。相反,SIRT1通过抑制过氧化物酶体增殖物激活受体-γ(PPARγ)和甾醇调节元件结合蛋白1(SREBP1)抑制脂肪生成。核SIRT6通过作为HIF1α的共同阻遏子和抑制糖酵解基因的表达来调节葡萄糖稳态。SIRT6还结合并直接去乙酰化肝X受体(LXR)和SREBP1,从而转录抑制脂肪生成。在胰腺β细胞中,SIRT6使FOXO1去乙酰化,触发其核输出并释放其对关键葡萄糖敏感基因(如葡萄糖转运蛋白2(GLUT2))的转录抑制。在线粒体中,SIRT3调节电子传递链复合物(ETC)内多个靶点的脱乙酰化。SIRT3与复合物I的NDUFA9亚基、复合物II的琥珀酸脱氢酶A(SDHA)和ATP合酶(复合物V)物理相互作用并调节其表达。作为对基质pH值降低和线粒体膜电位丧失的反应,SIRT3与ATP合成酶分离,导致基质蛋白快速脱乙酰基以重置线粒体乙酰基。SIRT3还可通过线粒体核糖体蛋白10(MRPL10)脱乙酰化调节呼吸复合物的转录,并通过调节三羧酸(TCA)循环(分别通过异柠檬酸脱氢酶2(IDH2)、乙酰辅酶A合成酶2(AceCs2)和谷氨酸脱氢酶(GDH)脱乙酰化)和刺激脂肪酸β-氧化和酮体生成(分别通过脱乙酰化长链酰基辅酶A脱氢酶(LCAD)和3-羟基-3-甲基戊二酰辅酶A合成酶2(HMGCS2))来协调代谢稳态。b、细胞内的能量供应受线粒体数量和形态的影响,SIRT1可直接去乙酰化PGC1α,促进线粒体生物合成,增加细胞器总数。同样,SIRT3和SIRT6分别通过AMP活化蛋白激酶(AMPK)和乙酰转移酶一般对照非抑制5蛋白(GCN5)间接激活PGC1α。新生成的线粒体通过融合过程被整合到线粒体网络中,融合过程由线粒体融合蛋白(Mfn1和Mfn2)和视神经萎缩蛋白1(Opa1)的自组装驱动。这些蛋白分别介导外膜和内膜的融合。融合诱导产生一个更相互连接的线粒体网络,从而稀释正常代谢过程中产生的活性氧(ROS)的毒性作用,有利于持续的能量生产。SIRT1和SIRT3分别通过直接去乙酰化Mfn2和Opa1促进线粒体融合。受损的线粒体通过分裂从健康网络中移除,分裂是由胞质动力相关蛋白1(Drp1)介导的。激活的Drp1通过与线粒体外膜上的线粒体分裂因子(MFF)结合而被招募到线粒体,并形成一个环状结构,切断细胞器。SIRT6通过Rho相关卷曲蛋白激酶1(ROCK1)阻止Drp1磷酸化,从而限制线粒体分裂。一旦分离,受损的线粒体失去膜电位(∆Ψ),并在外膜上积累丝氨酸/苏氨酸激酶PTEN诱导的假定激酶1(PINK1)。PINK1然后招募Parkin,一种胞质E3泛素连接酶,使外膜线粒体蛋白泛素化。泛素化被认为是招募自噬适配器LC3和GABARAP,导致线粒体吞噬。SIRT1和SIRT3可直接或间接诱导PINK1–Parkin依赖的有丝分裂吞噬,包括FOXO1和FOXO3的去乙酰化。SIRT1还可以通过降低哺乳动物雷帕霉素靶蛋白(mTOR)的表达来诱导线粒体吞噬,而SIRT3则通过激活AMPK来间接提高线粒体膜电位来增强线粒体吞噬。c、SIRTs在抗氧化途径中也有重要作用。SIRT1和SIRT6刺激转录因子核因子‑红细胞2相关因子2(Nrf2),Nrf2与抑制蛋白Keap1分离并诱导抗氧化基因的表达,包括硫氧还蛋白、硫氧还蛋白还原酶、硫氧还蛋白、过氧化物酶、谷胱甘肽过氧化物酶、超氧化物歧化酶1(SOD1)、过氧化氢酶(CAT)和几种谷胱甘肽S-转移酶。SIRT1、SIRT3和SIRT6还通过去乙酰化抑制NF-κB活化,从而限制促氧化基因的上调,包括NADPH氧化酶、黄嘌呤氧化还原酶、诱导型一氧化氮合酶、环氧化酶-2和细胞色素p450酶。此外,SIRT1和SIRT3通过促进下游靶基因如SOD2和CAT的表达,激活参与减少氧化应激的几个FOXO家族成员。转录后,SOD2易位到线粒体,其抗氧化活性由直接的SIRT3脱乙酰化调节。SIRT3还脱乙酰基酶IDH2,它促进抗氧化剂的再生并催化TCA循环的关键调节点。
线粒体动力学与生物发生
线粒体是一种普遍存在的细胞器,但其形态和数量在不同组织中差异很大,并且随着营养物质的供应而迅速变化。线粒体存在于动态网络中,其形态由分裂和融合的相反事件决定(图3b),严格控制以调节能量输出。
裂变使线粒体分裂,产生更小的线粒体,从而增加细胞器的总质量,并使能量供应迅速增加。然而,较小的线粒体增加了对膜去极化和氧化损伤的敏感性。分裂的分子机制包括细胞质动力相关蛋白1(dynamin-relatedprotein1,Drp1)向线粒体外膜的募集。Drp1结合线粒体分裂因子(mitochondrialfissionfactor,Mff),由此产生的复合物在线粒体周围形成环状结构,切断细胞器。
相反,融合诱导产生相互连接的线粒体网络,从而稀释正常代谢过程中产生的活性氧(reactiveoxygenspecies,ROS)的毒性作用,并有利于持续的能量生产。融合通过GTPases(即线粒体融合蛋白(Mfn1和Mfn2)和视神经萎缩1(opticatrophy1,Opa1))在线粒体内外膜上的自组装发生。SIRT1和SIRT3分别通过直接去乙酰化Mfn2促进线粒体融合和Opa1,而SIRT6通过Rho相关蛋白激酶1(Rho-associatedproteinkinase1,ROCK1)阻止Drp1磷酸化促进线粒体融合。
细胞内线粒体的数量也取决于生物发生和线粒体吞噬的相反过程。线粒体生物发生是新线粒体的新生,主要由PGC1α控制,SIRT1可通过直接去乙酰化PGC1α调控线粒体生物发生,而SIRT6可间接诱导PGC1α去乙酰化。SIRT3通过AMP活化蛋白激酶(AMP-activatedproteinkinase,AMPK)依赖的方式诱导PGC1α表达,从而促进线粒体生物发生。
线粒体自噬是一种质量控制过程,将受损的线粒体从健康的网络中分离出来,以供处理。SIRT1和SIRT3可直接诱导丝氨酸/苏氨酸激酶PTEN诱导的假定激酶1(PTEN-inducedputativekinase1,PINK1)–Parkin依赖的有丝分裂吞噬,间接通过FOXO1/3脱乙酰化等机制.SIRT1还通过降低哺乳动物雷帕霉素靶蛋白(mammaliantargetofrapamycin,mTOR)的表达诱导线粒体吞噬.而SIRT3通过激活AMPK增加线粒体膜电位间接增强线粒体吞噬功能。迄今为止,还没有报道SIRT6在线粒体吞噬中的作用。
氧化应激
与代谢调节相关的酶和亚细胞室通常是ROS的最大产生者,ROS最终被内源性抗氧化系统清除。当ROS产生超过细胞抗氧化酶的清除能力时,就会发生氧化损伤,并在触发不同器官的多种致病条件中起关键作用。
SIRTs协同作用,快速感知并增强细胞内的抗氧化防御(图3c)。SIRT1和SIRT6可激活转录因子核因子‑红细胞2相关因子2(nuclearerythroid2-relatedfactor2,Nrf2)与抑制蛋白Keap1分离,诱导抗氧化基因的表达。此外,SIRT1和SIRT3激活参与调节氧化应激的几个FOXO家族成员。FOXO1可通过调控下游靶基因如超氧化物歧化酶2(superoxidedismutase2,SOD2)和过氧化氢酶清除过量的ROS。此外,SIRT1、SIRT3和SIRT6脱乙酰化,从而抑制转录因子NF-κB的激活。这种机制限制了促进ROS产生的促氧化基因的上调。
DNA损伤与细胞凋亡
当抗氧化解毒系统不能将ROS维持在可耐受水平时,过多的细胞内促氧化环境会引发广泛且不可修复的DNA损伤,包括碱基修饰、DNA交联和单链或双链断裂,最终促进细胞死亡。核SIRT在保护DNA免受损伤方面具有保护作用。SIRT1和SIRT6对DNA修复的时空调控。K33处SIRT6的SIRT1脱乙酰化先于SIRT6聚合和识别双链DNA断裂,导致局部染色质重塑。此外,SIRT1通过Ku70脱乙酰基作用对抗DNA链断裂。
SIRT在细胞凋亡保护中也具有多方面的作用。在系膜细胞中,SIRT1通过激活p53和Smad7抑制细胞凋亡。SIRT1还通过FOXO4依赖性抑制Bim减少肾小球足细胞凋亡,Bim是启动内源性凋亡途径的关键决定因素。SIRT3通过控制ROS积累发挥抗凋亡作用,而SIRT6通过调节Notch信号和肌动蛋白细胞骨架保护细胞凋亡。
自噬
自噬是一个受严格调控的过程,通过溶酶体降解,使异常修饰的蛋白质、聚集体和受损的细胞器得以处置,从而使细胞免受凋亡。SIRTs通过直接去乙酰化自噬机制的必需蛋白以及间接机制参与自噬过程的调节。
饥饿激活SIRT1,SIRT1反过来使自噬成分去乙酰化,包括Atg5、Atg7和LC3。SIRT1还通过线粒体中Mfn2的去乙酰化激活自噬。现有数据表明,SIRT3和SIRT6可通过激活多种下游信号通路来调节自噬。SIRT3通过下调Notch-1–Hes-1通路增加肾小管细胞Beclin-1和LC3II的表达,从而促进自噬以及通过ERK–CREB信号通路。同样,SIRT6过表达通过减弱胰岛素样生长因子(insulin-likegrowthfactor,IGF)–Akt–mTOR信号通路诱导自噬;但其分子机制尚需进一步研究。
细胞衰老
细胞衰老是一种稳定的细胞周期阻滞形式,它是由时间老化过程中端粒缩短以及多种内源性和外源性刺激引起的。衰老是一个动态的、多步骤的过程,在此过程中,细胞经历基因表达的实质性改变,对促生长信号无反应,并形成复杂的衰老相关分泌表型(senescence-associatedsecretoryphenotype,SASP)。尽管仍存在争议,但越来越多的证据表明,sirt主要通过表观遗传调控基因转录、促进自噬和增强DNA损伤修复来减轻细胞衰老。
SIRT1可维持H3K9和H4K16乙酰化,从而抑制SASP促炎成分IL-6和IL-8的表达。此外,SIRT1通过微调ERK信号通路拮抗细胞衰老,调节LKB1及其下游靶蛋白AMPK的脱乙酰化。SIRT6与端粒结合,抑制SIRT6可诱导端粒损伤和衰老,与端粒H3K9高乙酰化和p53结合蛋白1(53BP1)结合有关。此外,SIRT6通过蛋白酶体降解细胞周期依赖性激酶抑制剂家族成员p27Kip1来限制细胞周期阻滞,从而延缓衰老。SIRT3通过与核膜蛋白和异染色质相关蛋白相互作用,最终稳定异染色质,从而对抗衰老,以及减轻氧化应激,促进自噬。
炎症与免疫反应
生物能量学中的一个新兴概念是细胞代谢与免疫细胞的招募和激活以及炎症的连续过程。虽然SIRT在炎症中作用的研究尚处于早期阶段,但越来越多的证据表明,在慢性炎症过程中,特定组织中NAD+水平和SIRT转录和/或蛋白水平持续降低。抑制NF-κB活化的能力使SIRT1在几种细胞类型中具有强烈的抗炎活性。SIRT1还通过高迁移率族蛋白B1(high-mobilitygroupbox1,HMGB1)脱乙酰基调节炎症,是一种高度保守的非组蛋白结构蛋白,通常定位于细胞核内,维持染色体的结构和功能。SIRT1的缺失增加HMGB1乙酰化并释放到细胞外空间,诱导导致全身炎症的信号级联反应。SIRT1和SIRT3的激活也具有抗炎作用,例如在急性炎症期间减少巨噬细胞和单核细胞产生肿瘤坏死因子(tumournecrosisfactor,TNF)。骨髓细胞特异性SIRT1缺失加重全身炎症实验模型的器官功能障碍和死亡率。SIRT3通过调节NLRP3炎症小体减少炎症,从而抑制氧化应激和促炎细胞因子。在肾结石小鼠模型中,SIRT3基因敲除导致肾内常驻B细胞减少,巨噬细胞和粒细胞浸润增加,提示SIRT3的表达影响组织中的免疫细胞景观。
2023年的一项研究确定了肾损伤过程中线粒体紊乱与炎症相关的机制。研究人员发现,线粒体RNA(mitochondrialRNA,mtRNA)的胞浆泄漏和胞浆模式识别受体视黄酸诱导基因1蛋白(retinoic-induciblegene1protein,RIG1,也称为DDX58)的激活导致炎症细胞的组织募集。RIG1样受体是病毒或细菌感染的关键感受器,RIG1依赖的胞质RNA感应通路的激活可诱导促炎状态。mtRNA诱导这种促炎信号通路活化的能力可能是线粒体内共生起源的结果,线粒体由细菌进化而来,与原核生物共享保守的结构基序。补充NAD+前体可恢复NAD+水平,改善线粒体功能,预防肾损伤。这一发现表明,SIRTs,特别是SIRT3,可能通过维持线粒体完整性和防止线粒体RNA泄漏而发挥保护作用。
Sirtuins在肾脏稳态中的作用
在健康个体中,肾脏是静息能量消耗最高的器官之一,其能量需求与其过滤血液、控制电解质平衡、维持酸碱平衡、重吸收营养和调节血压等生理作用相关。通过30多种终末分化细胞类型,发挥不同的功能,有完全不同的代谢要求,完成这些高度专业化的功能。
Sirtuins在肾脏发育中的作用
如果不了解肾脏胚胎学,就不能对肾脏细胞内在代谢的复杂性进行全面评估。在早期胚胎发生过程中,糖酵解流受到严格控制,并驱动细胞自我更新和增殖,因为最初的线粒体池在分裂的细胞中分离。尽管在胚胎中无氧代谢占优势,但线粒体成熟开始于肾发生中期,使氧化磷酸化能够满足与器官发育相关的增加的能量需求。从糖酵解到氧化代谢的转变是谱系特化的关键,以及决定胚胎分化过程中细胞命运的代谢驱动的表观遗传变化的结果。
在肾脏中,这种代谢转换的重要性通过发现代谢适应度不足影响肾单位祖细胞自我更新并影响每个肾脏的最终肾单位数来说明。SIRT3在胚胎肾脏中高度表达,需要抑制糖酵解和刺激线粒体功能来调节肾细胞前体池和行为,最终决定肾脏发生和出生时肾单位的赋能过程。在母体蛋白限制条件下,SIRT3表达降低与低肾单位数相关,是导致老年人肾功能不全易感性增加的主要原因。在母体蛋白限制的小鼠模型中,补充NAD+前体烟酰胺核糖可恢复SIRT3的表达和线粒体形态,并通过调节Opa1和SOD2的乙酰化来防止氧化应激,最终纠正肾单位赋能。对于其他SIRT在肾脏发育中的作用知之甚少。然而,SIRT1在体外与转录因子SIX2结合,提示SIRT1在控制肾祖细胞分化中具有潜在作用。
Sirtuins在肾脏生理中的作用
在成熟的肾脏中,不同的细胞类型依赖于不同的代谢途径来发挥其特定的功能。近端小管细胞富含线粒体,通过氧化磷酸化产生ATP,维持葡萄糖、离子和营养物质的主动运输。因此,线粒体SIRTs,特别是SIRT3和SIRT5,在调节近端肾小管细胞的正常稳态中具有重要作用。因为它们的缺失会影响ATP的产生并诱导线粒体碎片化。相反,足细胞利用糖酵解,通过细胞骨架重排和足突内裂孔膜的调节,快速产生调节过滤屏障所需的大量ATP。沉默调节糖酵解的SIRT6,已被证明可降低裂孔膜蛋白的表达。
SIRT1在集合管细胞中大量表达,通过抑制上皮钠通道(epithelialsodiumchannel,ENaC)α亚基的转录来调节钠平衡。在远端肾单位,SIRT1与促食欲肽胃饥饿素(ghrelin)协同作用,调节钠重吸收。SIRT7在不同的肾小管段表达,通过K+/Cl-共转运蛋白4(K+/Cl−cotransporter4,KCC4)的脱乙酰化调节酸碱和肾电解质的处理。这些结果表明SIRT1和SIRT7分别在调节肾脏的水盐平衡和酸中毒中起重要作用。
SIRT1也在调节肾血管腔室中起主要作用,因为它使内皮型一氧化氮合酶(othelialnitricoxidesynthase,eNOS)去乙酰化,产生一氧化氮,促进血管舒张。此外,SIRT1使缺氧诱导因子2α(hypoxia-induciblefactor2α,HIF2α)脱乙酰化,HIF2α在产生促红细胞生成素(erythropoietin,EPO)的间质细胞、内皮细胞和肾小球细胞中表达。这种去乙酰化导致EPO、氧调节基因和血管内皮生长因子(vascularothelialgrowthfactor,VEGF)的缺氧诱导。这些结果表明SIRT1在控制毛细血管密度和出生后血管适应方面具有重要作用。与野生型小鼠相比,Sirt3缺陷小鼠VEGF表达降低,HIF1α和血管紧张素-2表达上调,最终导致内皮损伤增加和肾毛细血管密度降低。总之,这些数据表明SIRTs在调节肾脏内环境平衡方面具有显著作用,对肾脏疾病的发展具有重要意义。

图4|Sirtuins在肾脏健康和疾病中的作用。a、Sirtuins(SIRTs)广泛表达于肾小球和肾小管。在肾小球中,SIRT1和SIRT6可维持足细胞的结构和功能完整性,从而维持滤过屏障的完整性。SIRT1和SIRT3在调控内皮细胞功能方面也起着重要作用。特别是,SIRT1通过调节内皮型一氧化氮合酶(eNOS)影响全身血压,而SIRT3通过调节血管内皮生长因子(VEGF)维持内皮完整性。SIRT1、SIRT3和SIRT5在近端小管中高度表达,在近端小管中它们保持线粒体功能完整性,使小管细胞产生大量的ATP,这些ATP是溶质重吸收所必需的。在远端小管中,SIRT1通过调节上皮钠通道(ENaC)的α亚基参与钠平衡和水重吸收的调控。SIRT7在近端和远端小管中表达,并通过K+/Cl−共转运蛋白4(KCC4)的去乙酰化调节酸碱和肾电解质处理。b、SIRT1、SIRT3、SIRT5、SIRT6和SIRT7在广泛的肾脏疾病中具有保护作用,包括促进能量平衡和线粒体功能,减少氧化应激、凋亡、炎症和纤维化。
Sirtuins与肾脏疾病
鉴于SIRTs在肾脏生理学中的重要作用,大量研究已经开始阐明其在影响肾脏的各种疾病中的作用。值得注意的是,SIRT1、SIRT3和SIRT6在急慢性肾脏疾病中的作用已被广泛研究(表2)。这些发现为寻找新的治疗方法以减轻肾脏疾病的进展铺平了道路。
表1|sirtuins在肾脏稳态中的作用
SIRT
部位
靶点
功能
参考文献。
SIRT1
足细胞
HIF2α
对缺氧的反应
116
内皮细胞
eNOS
血压控制
115
远端肾单位
生长激素释放肽(饥饿素)
钠重吸收的调节
112
集水管
ENaC
钠处理
111
SIRT3
小管细胞
Opa1、SOD2
线粒体稳态
100
内皮细胞
VEGF、HIF-1α和Angiopietin-2
血管完整性
118
SIRT5
小管细胞
MFN1、MFN2和OPA1
线粒体稳态
105
脂肪酸氧化
过氧化物酶体脂肪酸氧化的平衡
106
SIRT6
足细胞
Notch
裂孔膜完整性
110
SIRT7
小管细胞
KCC4
酸碱调节与肾脏电解质处理
113和,114
表2|sirtuins在肾脏疾病和衰老中的作用
Sirtuin
目标
活性
功能
参考文献。
SIRT1
PGC-1α
激活
刺激AKI的线粒体功能
122
NF-κB
抑制
抑制AKI炎症
124
Nrf2
激活
125
p65(NFkB),STAT3
抑制
减少DKD细胞凋亡
148
FOXO4
激活
65
Notch,HIF-2α,Smad3,Smad4,MMP7,
抑制
限制CKD的纤维化
168,169,211
MMP14
BMP2
抑制
抑制血管钙化
185
p53,Rb蛋白
抑制
促进ADPKD囊肿形成
189
组蛋白
抑制
狼疮性肾炎的抗体反应
192195年
FOXO,PGC-1α,PGC-1γ
激活
促进健康老龄化
198
SIRT3
Opa1、Mfn2、YME1L1
激活
刺激AKI的线粒体功能
48
SOD2
激活
增强AKI的抗氧化防御
131
GSK3β
激活
限制CKD的纤维化
175
NF-κB、TGF-β、Smad3
抑制
175
Opa1、Mfn2、,
激活
176
Drp1
抑制
176
KLF15
激活
179
PDHE1α
激活
阻碍CKD中的代谢重编程
177
AMPK
激活
抑制血管钙化
187
PGC-1α,AMPK,NAMPT
激活
促进健康老龄化
202
SIRT5
脂肪酸氧化
激活
保护AKI免受伤害
106
SIRT6
ERK1/2
抑制
抑制AKI炎症
129
Smad3
抑制
限制DKD的纤维化
153
β连环蛋白、HIPK2、TGF-β
抑制
限制CKD的纤维化
171172
Runx2
抑制
抑制血管钙化
184
NF-κB
抑制
促进健康老龄化
201
SIRT7
NF-κB
激活
AKI时促进肾小管损伤和肾脏炎症
140141
ADPKD,常染色体显性多囊肾病;AKI急性肾损伤;CKD慢性肾脏病;DKD糖尿病肾病。
急性肾损伤
无论最初的损伤如何,肾小管上皮细胞的功能障碍和丢失在急性肾损伤(acutekidneyinjury,AKI)的演变中起着关键作用。在这种情况下,线粒体功能障碍已被确定为肾小管细胞丢失之前最早的病理标志。与这一观察结果一致,越来越多的证据表明SIRT1对AKI具有保护作用。肾小管细胞特异性过表达SIRT1足以保护顺铂诱导的小鼠AKI。在AKI的缺血再灌注损伤(ischaemia–reperfusioninjury,IRI)模型中,需要依赖SIRT1的PGC1α去乙酰化来促进线粒体生物发生和氧化呼吸。维持损伤后小管修复的能量供应。与此结果一致,SIRT1沉默显著加重肾脏IRI。SIRT1保护肾脏免受损伤的能力也归因于其通过靶向NF-κB和Nrf2途径抑制肾小管上皮细胞炎症反应的作用。这些发现为使用SIRT1激活剂预防AKI提供了理论依据,包括黄酮类化合物、AMPK激动剂5-氨基咪唑-4-甲酰胺核糖核苷酸(AMPKagonist5-aminoimidazole-4-carboxamideribonucleotide,AICAR)和白藜芦醇。
SIRT6过表达已被证明可以减轻顺铂诱导的肾损伤。SIRT6通过去乙酰化组蛋白H3K9抑制ERK1和ERK2的表达,从而抑制炎症和凋亡。具有潜在临床意义的是,SIRT6在雌性而非雄性小鼠的受损肾脏中表达,这表明SIRT6可能是女性在AKI中保护作用的分子决定因素。
由于线粒体内稳态改变在AKI中占主导地位,线粒体SIRTs,特别是SIRT3的作用是一个非常有趣的话题。在顺铂诱导的AKI小鼠模型中,肾脏SIRT3蛋白表达降低与氧化应激标记物的积累以及Opa1高乙酰化和下调导致的线粒体分裂增加有关。已证明SIRT3通过ATP依赖性锌金属蛋白酶YME1L1的去乙酰化和失活来调节Opa1的表达,加速Opa1裂解,导致线粒体网络断裂。以类似的方式,SIRT3在肾脏IRI中保留Mfn2的泛素化和降解。这些数据表明,缺乏SIRT3导致的线粒体动力学向碎片化的失衡是AKI各种实验模型的一个统一特征,导致线粒体通透性、ATP耗竭、ROS过度产生和凋亡因子释放。SIRT3在AKI中的非冗余作用得到证实,与野生型小鼠相比,SIRT3缺陷小鼠在顺铂给药后表现出更严重的疾病和过早死亡。这些研究的转归是SIRT3是药物干预缓解AKI的潜在靶点。AICAR可恢复野生型小鼠SIRT3的表达和活性,改善肾功能,减轻肾小管损伤,但在SIRT3基因敲除小鼠中无此作用。其他潜在的SIRT3激活剂,如姜黄素,黄酮类化合物,白藜芦醇,水飞蓟宾和锡钙素对实验性AKI也有保护作用。
恢复和激活SIRT3的替代策略是NAD+补充。在小鼠模型中,补充NAD+前体烟酰胺单核苷酸(nicotinamidemononucleotide,NAMN)和烟酰胺核苷(nicotinamideriboside,NR)对AKI有保护作用。顺铂诱导肾小管上皮细胞AKI时NAD+生物利用度显著降低,从而对SIRT3活性产生负面影响。人脐带间充质干细胞(umbilicalcord-derivedmesenchymalstemcells,UC-MSCs)与顺铂损伤的肾小管细胞共孵育诱导NAD+生物合成途径中几个关键酶的上调,包括在挽救途径中起作用的烟酰胺磷酸核糖转移酶(nicotinamidephosphoribosyltransferase,NAMPT)和在从头途径中起作用的犬尿氨酸酶(图2c)。这些酶的上调伴随着顺铂治疗小鼠肾组织中NAD+水平和SIRT3活性的恢复。研究人员还发现,UC-MSCs增强SIRT3活性,使健康线粒体通过富含微管蛋白的投射在肾小管上皮细胞之间进行细胞间转移,有利于受损肾小管细胞之间的生物能量串扰。这些作用转化为UC-MSCs通过NAMPT和PGC1α上调SIRT3对AKI小鼠肾功能和线粒体稳态的保护作用。其他SIRTs的保护作用相反,两项小鼠研究表明SIRT7在AKI过程中具有有害作用,但其病理机制尚未明确。
与此相反,SIRT1在自身免疫性肾病和多囊肾病中可促进自身抗体产生和囊肿形成,而SIRT7在急性肾损伤中可诱导炎症反应。
关于SIRTs在AKI患者中的作用知之甚少。然而,据报道,在AKI高危住院患者中NAD+生物合成受损。这一发现表明,使用增加NAD+水平的治疗方法,类似于在实验模型中研究的方法,对AKI患者可能是一种有价值的肾脏保护策略。
糖尿病肾病
糖尿病肾病(Diabetickidneydisease,DKD)是世界范围内导致肾功能衰竭的主要原因,也是心血管疾病的危险因素。SIRTs由于其在调节细胞代谢中的突出作用而在DKD中被广泛研究。
SIRT1激动剂可以改善代谢参数,如糖耐量和胰岛素抵抗,延长糖尿病动物的存活时间。除了这些积极的代谢作用外,SIRT1还直接限制足细胞损伤。在DKD小鼠中,SIRT1的条件性足细胞缺失增加蛋白尿并加重肾损害。SIRT1的保护作用与足细胞内STAT3和NF-κBp65亚基乙酰化增加有关。此外,SIRT1表达降低可导致DKD小鼠肾小球FOXO4高乙酰化和促凋亡因子的诱导。在早期DKD小鼠模型中短期应用NAD+前体NAMN通过上调SIRT1和NAD+挽救途径实现肾脏保护。与此证据一致,SIRT1在糖尿病患者肾小球中下调,旨在增加SIRT1的饮食干预降低了DKD患者晚期糖基化终末产物水平并恢复了抗氧化防御。
糖尿病小鼠SIRT6表达和AMPK去磷酸化水平降低,伴有线粒体形态异常和进行性肾损伤。在高糖暴露的大鼠系膜细胞(DKD的体外模型)中,SIRT6被miR-33a-5p依赖的通路下调。在链脲佐菌素(streptozotocin,STZ)诱导的糖尿病小鼠中,circRNAE3泛素蛋白连接酶(circRNAcircITCH)通过抑制miR-33a-5p从而增加SIRT6的表达来改善肾脏炎症和纤维化。SIRT6可通过直接结合Smad3并使其脱乙酰化来调节纤维化,阻止其核积聚和与上皮-间质转化和肾纤维化相关基因的转录活性。在STZ诱导的糖尿病小鼠中,SIRT6的管状细胞特异性缺失通过增加基质金属蛋白酶Timp1的表达和细胞外基质沉积增强纤维化表型。SIRT6过表达还可通过使巨噬细胞极化至保护性M2表型并限制白细胞介素和细胞因子的产生而保护STZ诱导的DKD大鼠的足细胞损伤。提示SIRT6在DKD中具有调节免疫应答和保护肾脏免受炎症损伤的作用。SIRT6在DKD患者足细胞中的表达也受到抑制。
SIRT3在DKD中也有保护作用。肾小管细胞中SIRT3的过表达通过上调Akt–FoxO信号通路抑制ROS的积累,从而对抗高血糖介导的肾细胞凋亡。SIRT3在小鼠和DKD患者肾脏中的表达降低。和厚朴酚可选择性激活SIRT3,减轻蛋白尿,改善糖尿病小鼠模型(BTBRob/ob小鼠)肾小球损伤。这种肾脏保护作用依赖于和厚朴酚诱导的SIRT3激活,SIRT3激活通过激活SOD2和恢复肾小球细胞PGC1α表达来保护线粒体功能和超微结构。
另一方面,Sirt3的缺乏会增加高脂饮食引起的小鼠肾脏疾病的严重程度,这种情况类似于人类的代谢综合征。在这种情况下,Sirt3缺乏导致脂质积累和线粒体损伤的肾小管细胞。肾小管异位脂质积聚是DKD患者的一个显著病理特征,其发生是由于骨骼肌和脂肪组织产生的激素流星蛋白样蛋白(meteorin-likeprotein,Metrnl)水平降低所致。Metrnl可激活SIRT3-AMPK信号轴,维持线粒体稳态,促进产热,从而减轻脂质积聚。应用重组Metrnl可减少糖尿病小鼠的脂质积累并改善肾脏损伤,提示SIRT3可通过一种新机制延缓DKD的进展。
慢性肾脏病与纤维化
无论最初的病因是什么,几乎所有进展型慢性肾脏病(chronickidneydisease,CKD)的最终结果都是:广泛纤维化是主要的病理现象。在CKD大鼠模型中,NAD+生物合成显著下调,表明SIRTs的活性也受到损害,包括与其抗纤维化机制相关的活性。
据报道,SIRT1在局灶节段性肾小球硬化患者肾活检标本中的表达降低。沉默SIRT1可加重单侧输尿管梗阻(unilateralureteralobstruction,UUO)小鼠的纤维化,而通过药物干预或基因操作刺激SIRT1可在进行性纤维化肾病的实验模型中发挥抗炎作用并减少基质蛋白积累。SIRT1的抗纤维化活性与其通过调节Smad蛋白抑制TGFβ信号通路的能力有关。SIRT1使Smad3和Smad4脱乙酰化,从而损害促纤维化基因的转录,包括IV型胶原、纤维连接蛋白和基质金属蛋白酶7(matrixmetalloproteinase7,MMP7)。SIRT1的额外抗纤维化作用归因于其调节内皮细胞功能的能力。在小鼠中,靶向缺失SIRT1在内皮中,由于基质金属蛋白酶14(matrixmetalloproteinase14,MMP14)的下调、HIF2α–Notch1轴的刺激和内皮糖萼的蛋白水解片段的释放,导致血管舒张受损并促进纤维化。
SIRT6也被证明对肾间质纤维化有保护作用。在小鼠中,SIRT6在纤维化损伤后上调,并与β-连环素相互作用。由此产生的复合物与成纤维β-连环素靶基因的启动子结合,并通过SIRT6依赖的组蛋白去乙酰化阻止其转录。此外,SIRT6过表达通过下调同源结构域相互作用蛋白激酶2(homeodomaininteractingproteinkinase2,HIPK2)来预防高腺嘌呤饮食喂养的小鼠肾间质纤维化,HIPK2作用于各种促纤维化途径的上游。在肾小管细胞中,糖原合成酶激酶3β(glycogensynthasekinase3β,GSK3β)磷酸化SIRT6可阻止其蛋白酶体降解,从而产生抗纤维化作用。
线粒体是包括肾脏在内的器官纤维化的关键因素。SIRT3已被证明具有抗心脏纤维化作用,但对其在肾纤维化中的作用知之甚少。在Sirt3缺乏小鼠中,年龄依赖性肾纤维化增加,可能是由于TGFβ信号和GSK3β高乙酰化增加,最终导致Smad3激活。Sirt3缺陷小鼠的Opa1和Mfn1水平降低,Drp1水平升高,导致线粒体分裂,线粒体分裂与肾功能不全和纤维化相关。另一项研究发现,在肾纤维化早期,SIRT3表达降低伴随着肾小管细胞线粒体乙酰化增加。对UUO模型肾小管细胞的乙酰组分析表明,SIRT3介导的赖氨酸385处丙酮酸脱氢酶E1α(pyruvatedehydrogenaseE1α,PDHE1α)脱乙酰基在与肾纤维化相关的代谢重编程中起关键作用。和厚朴酚激活SIRT3通过调节线粒体动力学和NF-κB–TGF-β1–Smad信号通路预防UUO诱导的肾纤维化。SIRT3还显示使足细胞中KLF15去乙酰化,KLF15是细胞外基质蛋白合成的负调节因子。在具有纤维化表型的小鼠中,SIRT3在内皮细胞中的过表达对糖尿病相关肾纤维化的保护。相反,内皮细胞中Sirt3缺乏可诱导代谢重编程,从而刺激肾小管上皮细胞中TGFβ–Smad3依赖的间充质转化。
钠-葡萄糖共转运蛋白2(Sodium-glucosecotransporter2,SGLT2)抑制剂是一种有效的治疗方法,可减少人类CKD的进展。据报道,SGLT2抑制剂卡格列嗪可恢复SIRT3表达并改善高血压肾损伤大鼠的上皮-间质转化。SGLT2抑制剂诱导有益作用的特征模式,包括减少氧化应激、恢复线粒体健康、增强线粒体生物发生、减少促炎和促纤维化途径以及保持细胞和器官完整性和活性。SGLT2抑制剂的肾脏保护作用可通过特异性抑制自噬和AMPK而被消除,提示SIRT3在这些保护机制中可能发挥作用。
血管钙化
血管钙化发生在血管系统的内膜和中层,其特征是肌肉动脉壁增厚和失去弹性。血管钙化的高患病率导致CKD患者的心血管发病率和死亡率。CKD血管钙化的机制尚不完全清楚,靶向治疗的发展也不令人满意。新的数据表明SIRTs可能在血管钙化的发病机制中起主要作用。
据报道,SIRT1在老龄小鼠主动脉中下调,导致SASP因子子集的表达,包括促成骨分子骨形态发生蛋白2(bonemorphogeneticprotein2,BMP2)和促进血管平滑肌细胞(vascularsmoothmusclecell,VSMC)衰老。在CKD大鼠模型中,天然多胺亚精胺上调SIRT1并减轻血管钙化。
SIRT6在CKD和血管钙化患者的桡动脉组织中显著下调。此外,VSMCs中SIRT6的缺失加剧了CKD小鼠的血管钙化。SIRT6结合并去乙酰化runt相关转录因子2(deacetylatesrunt-relatedtranscriptionfactor2,Runx2),Runx2在成骨细胞分化和骨骼形态发生中发挥作用,加速其降解并限制VSMCs的成骨转分化。
在CKD大鼠模型中,SIRT3蛋白水平升高可改善线粒体功能,抑制线粒体氧化应激,限制VSMCs钙化。这些保护作用可能与SIRT3调节AMPK信号187的能力有关。
多囊肾病
常染色体显性多囊肾病(Autosomal-dominantpolycystickidneydisease,ADPKD)是一种遗传性疾病,由多囊蛋白-1(polycystin-1,PKD1)或多囊蛋白-2(polycystin-2,PKD2)突变引起,约400–1000人中有1人受影响。ADPKD的特点是多发性双侧肾囊肿取代正常肾组织,导致肾衰竭。
SIRTs在ADPKD中的潜在致病作用尚处于早期阶段。然而,沉默或药物抑制SIRT1已被证明可以减轻小鼠中囊肿的形成。ADPKD中SIRT1的有害作用归因于其抑制视网膜母细胞瘤肿瘤抑制蛋白(Rb)和p53的功能,从而促进上皮细胞持续生长和囊肿形成。基于这些发现,对ADPKD患者进行了口服烟酰胺(高剂量抑制SIRTs)的疗效试验。研究表明,烟酰胺膳食补充剂的安全性和耐受性,但没有有益的效果;烟酰胺组和安慰剂组在治疗12个月后,身高调整后的总肾体积变化没有差异。
自身免疫疾病
SIRTs在自身免疫性疾病中的潜在作用是一个新兴领域,目前可获得的数据很少。然而,临床前和临床研究表明SIRT1在各种自身免疫性疾病的发病机制中发挥作用。活动性狼疮性肾炎患者尿液中SIRT1mRNA和蛋白水平显著高于缓解期患者和健康人。此外,SIRT1表达与有无肾炎的系统性红斑狼疮患者的抗双链DNA(anti-double-strandedDNA,dsDNA)抗体密切相关。一项研究分析了367例SLE患者和290名健康人的外周血样本,发现SIRT1启动子变异体rs3758391改变了疾病发病率,rs3758391T等位基因是狼疮性肾炎的危险因素。在易发生狼疮性肾炎的MRL/lpr小鼠中,给予SIRT1短干扰RNA(short-interferingRNA,siRNA)可减少病理损伤、抗dsDNA抗体水平和肾IgG沉积。SIRT1在幼稚B细胞中高表达,并可通过去乙酰化组蛋白和非组蛋白表观遗传调节抗体反应。
肾脏老化
肾脏对衰老过程高度敏感,随着年龄的增长,肾脏更容易发生急性和慢性损伤。临床前研究发现,肾脏SIRT1活性随着年龄的增加而降低,同时细胞内NAD+池丢失,导致线粒体肿胀和嵴破坏增加。热量限制改善了Sirt1有能力老年小鼠,但没有改善Sirt1缺陷老年小鼠的线粒体异常,并且足细胞中Sirt1的靶向缺失加速了老年小鼠的肾损伤和细胞衰老。同样,Sirt1基因敲除可导致内皮细胞过早衰老,而Sirt1激活剂或NAD+前体烟酰胺单核苷酸给药可增加小鼠在衰老过程中的肾弹性。热量限制还可增加老年小鼠SIRT6表达,减弱NF-κB信号,改善肾功能。关于SIRT3,缺乏血管紧张素Ⅱ型受体(angiotensintypeIAreceptor,AT1AR)的小鼠的促长寿表型与Nampt和Sirt3在肾小管细胞中的表达,与线粒体密度增加和氧化应激减少平行。
表3|具有良好特性的sirtuin激活剂和抑制剂
化合物
实验性肾脏疾病的生物学效应
参考文献
天然STACs
白藜芦醇
刺激SIRT1并预防AKI
128
通过刺激SIRT1和SIRT3限制AKI、DKD、CKD和膜性肾病的肾脏损害
137
黄酮类化合物
刺激SIRT1预防AKI
126
通过SIRT1依赖性激活Nrf2和抑制NF-κB信号通路在AKI中发挥肾脏保护作用
135
水飞蓟宾
激活SIRT3,改善线粒体功能和生物能量学,改善AKI期间的肾损伤
136
锡钙素
通过AMPK刺激SIRT3并限制AKI期间的肾损伤
138
和厚朴酚
刺激SIRT3减轻2型糖尿病BTBR-ob/ob小鼠蛋白尿、肾小球损伤和足细胞损伤
157
UUO模型中通过调节线粒体动力学和NF-κB–TGF-β1–Smad通路激活SIRT3并预防肾纤维化
177178
亚精胺
上调SIRT1抑制CKD大鼠血管钙化
186
NAD+促进化合物
外源性NAD+
维持SIRT1和SIRT3活性并保护高糖诱导的系膜肥大
212
烟酰胺核糖
在母体蛋白限制的情况下刺激SIRT3的表达和活性并促进正常肾发生
101
防止线粒体RNA泄漏并限制AKI期间的炎症
92
AICAR
刺激SIRT1并预防AKI
127
通过激活NAMPT、PGC1-a和SIRT3刺激AMPK并维持AKI线粒体稳态
48
UC-MSCs
通过NAMPT和KYNU刺激NAD+产生维持AKI线粒体内稳态
139
NAMN
防止线粒体RNA泄漏并限制AKI期间的炎症
92
通过上调SIRT1和NAD+挽救途径在早期DKD中发挥肾脏保护作用
149
合成STACs
SRT1720
在UUO模型中刺激SIRT1并限制纤维化
213
SRT2104
通过SIRT1刺激抗氧化反应,延长寿命并改善小鼠的整体健康
214
SRT2183
刺激SIRT1减少UUO模型肾髓质纤维化和凋亡
164
SRT3025
刺激SIRT1并减轻蛋白尿和GFR下降,从而减少残余肾模型中的肾小球硬化和肾小管间质纤维化
163
AKI急性肾损伤;CKD慢性肾脏病;DKD糖尿病肾病;GFR肾小球滤过率;mtRNA,线粒体RNA;STAC,sirtuin激活化合物;T2D2型糖尿病;UUO单侧输尿管梗阻。
靶向sirtuins
鉴于SIRTs所赋予的深远健康益处,发现sirtuin激活化合物(sirtuin-activatingcompounds,STACs)是该领域的主要目标(表3)。第一个鉴定的STAC是白藜芦醇,它被认为是通过作为变构调节剂激活SIRT1,并降低NAD+和乙酰化肽的KM,尽管这种作用机制有争议。近200项白藜芦醇临床试验处于不同的完成阶段。在大多数白藜芦醇已被检测的疾病中,该治疗具有中性效果。这种缺乏保护的主要原因是白藜芦醇的生物利用度有限,这是该化合物治疗应用的主要障碍。为了克服这一限制,合成的小分子已经被开发出来以比白藜芦醇更高的亲和力刺激SIRTs。然而,这种方法具有挑战性,因为酶抑制剂通常比激活剂更容易开发。酶抑制剂通常通过直接干扰催化位点发挥作用,而激活剂需要一个不同于底物的变构结合位点才能正常发挥作用。迄今为止,四种合成的STAC,SRT1720、SRT2104、SRT2183和SRT3025,已在各种实验模型中显示可增加SIRT1活性并限制肾损伤,SRT2104和SRT3025已进入临床试验。迄今为止,大多数这些试验都取得了中性结果。然而,一些SRT2104的试验报告了有益于2型糖尿病患者的血脂谱、银屑病患者的皮肤组织学以及健康个体的脂多糖诱导的炎症和凝血。合成STACs出现了一些安全问题,例如,据报道,SRT3025可诱发潜在致命的促心律失常。
增加SIRTs酶活性的另一种方法是补充提高NAD+水平的小分子,例如外源NAD+或其前体。还可以通过阻断NAD+降解来增加NAD+生物利用度,例如通过抑制NADaseCD38。CD38抑制剂TNB-738同时结合两个不同的CD38表位,在体外可有效抑制CD38酶活性,导致细胞内NAD+水平和SIRT活性增加。临床试验的可用数据表明,针对NAD的治疗是安全的。然而,这种方法缺乏SIRT异构体选择性,这可能会妨碍其作为治疗性化合物的适用性。
结论和未来展望
自从SIRTs的首次发现以来,对该蛋白家族的理解变得越来越全面。虽然最初的研究集中于确定SIRT酶活性的关键底物,但后来的研究支持这样的观点,即SIRT调节靶蛋白的功能簇,以协调各种细胞过程中的协调生理反应,包括代谢、氧化应激、细胞存活和基因组稳定性。现有数据表明,SIRTs对于平衡包括肾脏在内的代谢活跃器官中过多的应激源至关重要,影响生理学和病理生理学。增加对SIRT在各种肾细胞类型中对代谢和非代谢途径的复杂调控的理解可能提供一个独特的机会,以确定治疗广泛肾脏疾病的新靶点。靶向SIRT的干预措施在治疗肾脏疾病和对抗年龄相关性肾脏疾病方面具有巨大潜力,STACs在实验模型中的强大作用证明了这一点。遗憾的是,在临床前研究中的巨大努力只产生了临床研究中的少数小分子。由于缺乏针对单个SIRT亚型的选择性候选化合物以及现有候选化合物的中等效力、有限的生物利用度和较差的药代动力学和药效学特征,SIRT调节器从试验台到临床的转化受到阻碍。SRT2104在代谢紊乱、银屑病和炎症中的早期临床数据为新型SIRT激活剂的继续研究提供了有力的支持。在未来,这种SIRT靶向治疗有可能用于增加人类的平均健康跨度。
阅读原文2024



